


What is Arbor?
Arbor is a web-based software package for phylogenetic comparative analyses across the tree of life. Together with Luke Harmon, Bob Thacker, Curt Lisle and Jeff Baumes we continue to develop the analytical and visualization tools to enable researchers to investigate their biodiversity data in a phylogenetic and biogeographic context. Our main website is:
What will Arbor be able to do for me?
To start out with, Arbor will be able to do the most common comparative analyses, including:
- Phylogenetic signal
- Ancestral state reconstruction
- Independent contrasts
- Comparative model fitting (e.g. Mk, threshold, and continuous character models)
- PGLS
- Fitting Birth-death models
- Community phylogenetics
- BiSSE / MuSSE / QuaSSE / GeoSSE
- Analyses of cospeciation
You can also visualize trees and maps simultaneously to investigate trait distribution over phylogenetic and spatial scales.
We are actively building up towards more complicated analyses and more intuitive visualization.
What is an Arbor workflow?
Phylogenetic trees are increasingly available, and can be used for a wide range of analyses, including studies of speciation, trait evolution, range size evolution, and many other things. Phylogenetic comparative methods have been developing rapidly in recent years. Most analyses are currently done in R, and a number of useful packages are available (e.g. ape, diversitree, geiger, caper, phytools, and others). Arbor seeks to enable end-users to carry out these analyses by building workflows.
Building an Arbor workflow will have more in common with making something out of legos than with computer programming. For example, the figure below shows a mockup of an Arbor workflow to calculate phylogenetic independent contrasts (PICs). End users will import a phylogenetic tree and some traits data – perhaps from their local computer, or maybe from a remote source like Open Tree of Life or Encyclopedia of Life. The workflow below then allows the user to connect a few steps together to calculate PICs and use them to test for a correlation between two traits.
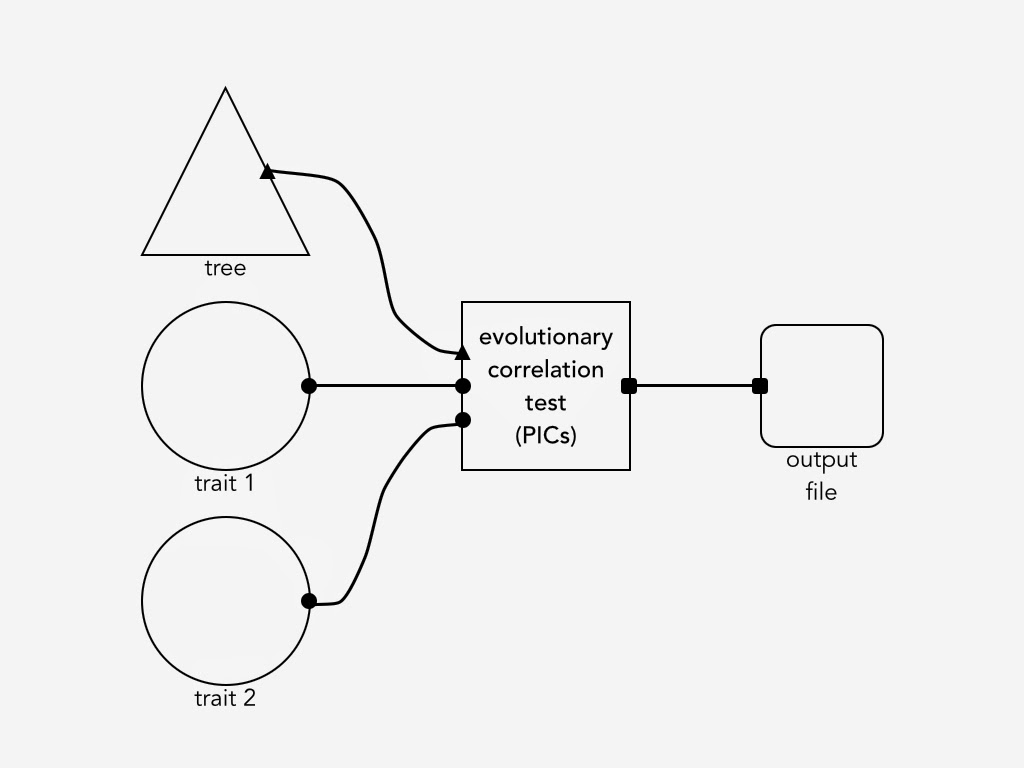
A sample workflow for calculating phylogenetic independent contrasts (PICs) to test for an evolutionary correlation between two traits.
The PIC workflow above is simple, but Arbor will enable more complex analyses as well. For example, imagine you are interested in analyzing geographic range evolution in your group of species.
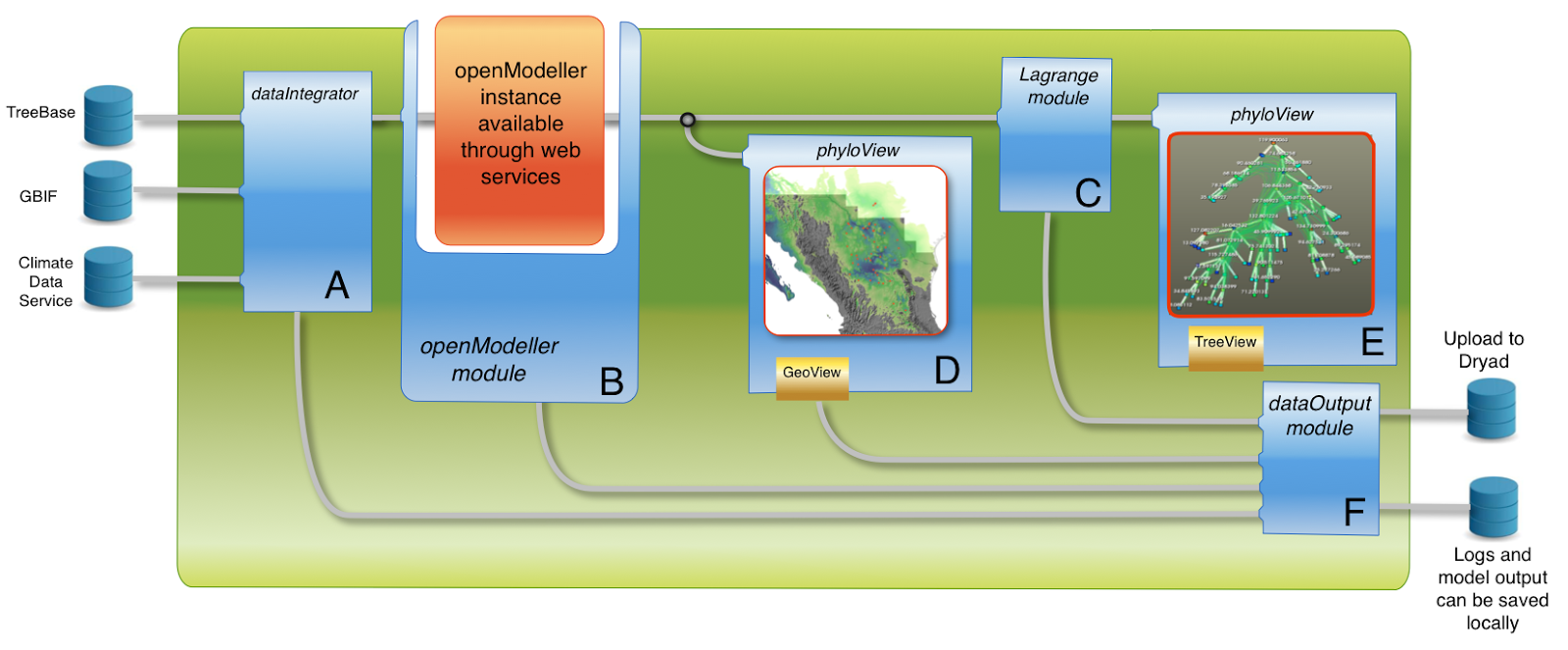
A more complex workflow that uses data from web services to combine ecological niche models, Lagrange, and phylogenetic comparative analyses.
The workflow above shows how you might use Arbor to link phylogenetic, trait, and geographic data together in a comprehensive set of comparative analyses. If you want to get an idea of other examples of workflow software in biology, you can check out other examples like Galaxy and the iPlant Discovery Environment.
How can I participate in the project?
You can follow our blog, visit our main site, and find our code on github. We will also be hosting a series of hackathons to be announced at a later date.
http://www.arborworkflows.com/
You can go here to find all functions.
You can also go here to check out the tutorials.
And here is a getting started page that includes some video tips for loading data and running analyses.